In recent years, the continuous advancements in sequencing technology have markedly refined genome sequencing techniques, yielding significant achievements in both animal and plant genomic research. Numerous plant genome drafts and detailed maps have emerged, supplying invaluable resources for the scientific community. This article provides an in-depth analysis of the characteristics innate to third-generation sequencing technologies and systematically reviews the progress in pre-sequencing preparations, genome assembly, annotation processes, and comparative genomics. Furthermore, it elucidates the unique features and challenges inherent to plant genome research. Through comprehensive plant genome sequencing, researchers can not only obtain the genome sequences and key functional genes of plants, thus supporting in-depth molecular investigations into plant evolution, gene composition, and regulatory mechanisms, but also offer essential reference value and guidance for forthcoming plant genomic studies.
The sequencing of entire plant genomes constitutes a highly influential and extensive endeavor, facilitated by advanced genomic technologies. This initiative aims to elucidate the genetic blueprints of numerous essential plant species. Moreover, this project enables precise analysis of genetic variability and mutations at the population level, thereby establishing a robust foundation for genomic-level plant research. Consequently, it offers invaluable guidance and support for traditional research paradigms.
Over the past two decades, significant advancements have been achieved in the field of whole-genome sequencing for both animals and plants. The initiation of the Human Genome Project (HGP) in 1990 marked the advent of large-scale genomic DNA sequencing. By the year 2000, the preliminary completion of the human genome draft indicated that extensive DNA sequencing had become a routine methodological approach. However, comparative to animal genomics, the study of plant genomes presents distinct challenges. Plant genomes are often characterized by polyploidy, considerable genome size, high heterozygosity, and the presence of extensive repetitive sequences and entirely or partially duplicated genome segments. Consequently, it was virtually impossible to sequence certain complex plant genomes using traditional Sanger sequencing or early second-generation sequencing technologies.
With the continuous advancements in sequencing technologies and the gradual reduction in associated costs, an increasing number of plant genome sequencing projects have been initiated and have yielded substantial results. The publication of the complete genome sequence of the model organism Arabidopsis thaliana in 2000 marked the commencement of comprehensive plant genome research. Subsequently, the completion of the rice (Oryza sativa) genome sequence in 2002, the first among cereal crops, established a crucial foundation for the exploration of gene annotation and the study of orthologous genes in other plant species. In-depth analyses of these genomic datasets have enhanced the understanding of critical issues pertaining to species growth, development, evolution, and origin. Moreover, these studies have expedited the discovery of novel genes and the process of species improvement, thereby paving the way for genome sequencing efforts in other plant taxa.
Over the past decade, genomic research on numerous plant species, including Populus (poplar), Vitis vinifera (grape), Sorghum bicolor (sorghum), Zea mays (maize), Cucumis sativus (cucumber), Glycine max (soybean), Ricinus communis (castor bean), Malus domestica (apple), Fragaria vesca (strawberry), Theobroma cacao (cocoa tree), Brassica rapa (Chinese cabbage), and Solanum tuberosum (potato), has been documented. These advancements have been facilitated by the rapid evolution and widespread application of various sequencing technologies, which have substantially shortened the time required for whole-genome sequencing and reduced associated costs. Concurrently, these studies have refined research objectives and accelerated experimental design processes. Consequently, the understanding of physiological and biochemical mechanisms in plant growth and development has been elevated to the molecular level, providing novel perspectives for comprehending gene structure, composition, function, regulation, and species evolution at the molecular level.
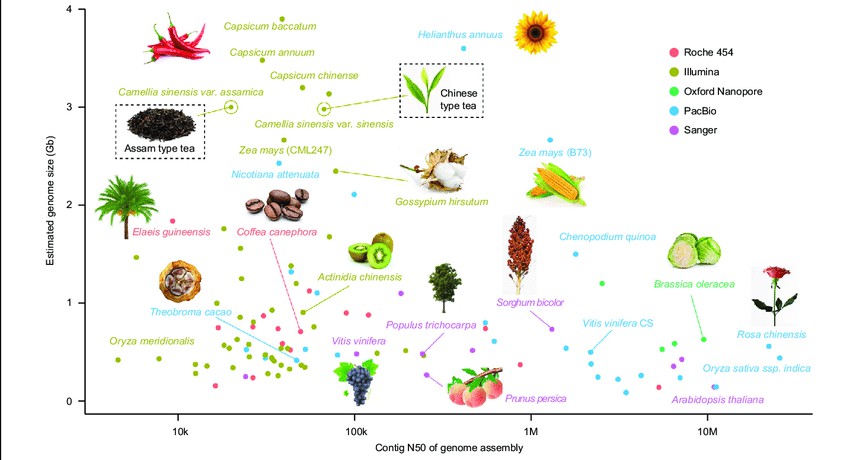
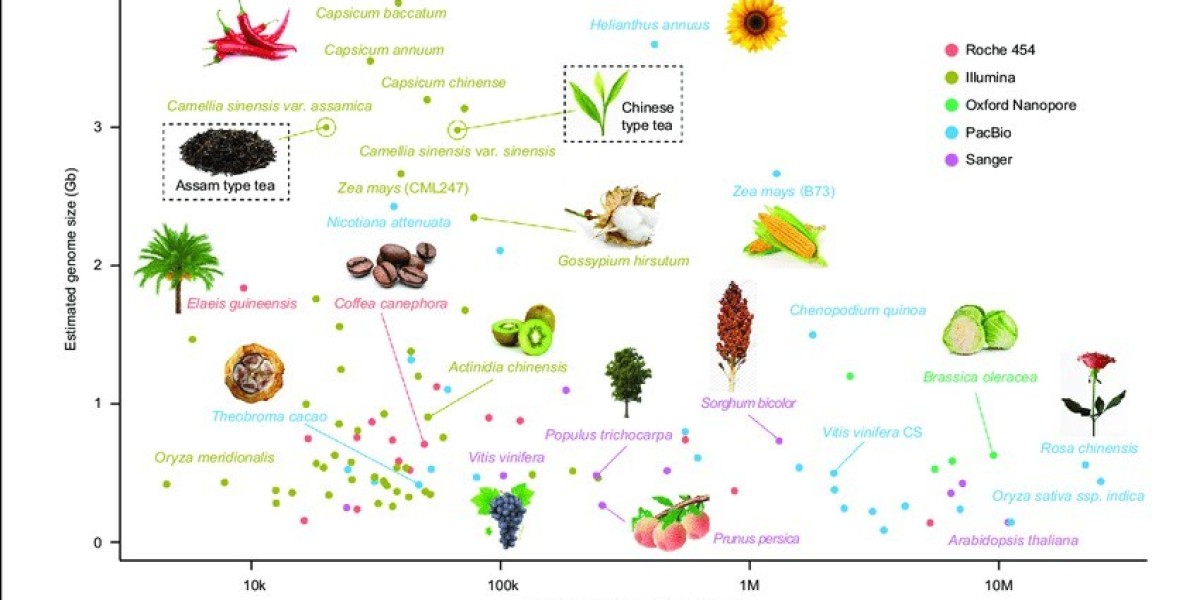
Figure 1 illustrates the current progress in genome sequencing of various plant species. The x-axis represents the contig N50 of the genome assembly, while the y-axis displays the estimated genome size for each plant. Different sequencing platforms are denoted by colors: red for Roche 454, brown for Illumina, green for Oxford nanopore, blue for PacBio SMRT, and pink for Sanger. Tea plants are highlighted with a rectangular box (Xia et al., 2020).
Table 1: The Partial Published Complete Plant Genome Sequencing
| Plant Name (Scientific Name) | Genome Size | Family, Genus | Sequencing Platform |
|---|---|---|---|
| Arabidopsis thaliana | 125M | Brassicaceae, Arabidopsis | Sanger construct BAC/TAC library |
| Oryza sativa | 466M | Poaceae, Oryza | Sanger whole-genome shotgun |
| Populus trichocarpa | 480M | Salicaceae, Populus | Sanger whole-genome shotgun |
| Chlamydomonas reinhardtii | 130M | Chlamydomonadaceae | Sanger whole-genome shotgun |
| Vitis vinifera | 490M | Vitaceae, Vitis | Sanger whole-genome shotgun |
| Carica papaya | 370M | Caricaceae, Carica | Sanger whole-genome shotgun |
| Sorghum bicolor | 730M | Poaceae, Sorghum | Sanger whole-genome shotgun |
| Zea mays | 2300M | Poaceae, Zea | Sanger clone-by-clone |
| Cucumis sativus | 350M | Cucurbitaceae, Cucumis | Sanger + Illumina GA |
| Glycine max | 1100M | Fabaceae, Glycine | Sanger whole-genome shotgun |
| Brachypodium distachyon | 260M | Poaceae, Brachypodium | Sanger whole-genome shotgun |
| Ricinus communis | 350M | Euphorbiaceae, Ricinus | Sanger whole-genome shotgun |
| Malus domestica | 742M | Rosaceae, Malus | Sanger + 454 sequencer |
| Fragaria vesca | 240M | Rosaceae, Fragaria | Roche/454, Illumina/Solexa |
| Theobroma cacao | 430M | Malvaceae, Theobroma | Illumina whole-genome shotgun |
| Solanum tuberosum | 844M | Solanaceae, Solanum | Illumina, 454 whole-genome shotgun |
| Brassica rapa | 485M | Brassicaceae, Brassica | Illumina GA |
| Cannabis sativa | 534M | Cannabaceae, Cannabis | Illumina HiSeq, 454 |
| Juglans regia | 667M | Juglandaceae, Juglans | Illumina GA, HiSeq 2000 |
| Setaria italica | 423M | Poaceae, Setaria | Illumina HiSeq 2000 |
| Prunus armeniaca | 280M | Rosaceae, Prunus | Illumina GA |
| Citrus sinensis | 367M | Rutaceae, Citrus | Illumina GAⅡ, WGS |
| Citrullus lanatus | 425M | Cucurbitaceae, Citrullus | Illumina |
| Hordeum vulgare | 5.1G | Poaceae, Hordeum | Illumina + Roche 454 |
| Phyllostachys edulis | 2.05G | Poaceae, Phyllostachys | Illumina |
| Triticum aestivum | 4.94G | Poaceae, Triticum | Illumina HiSeq |
| Picea abies | 19.6G | Pinaceae, Picea | Whole-genome shotgun |
| Nelumbo nucifera | 879M | Nelumbonaceae, Nelumbo | Illumina, 454 |
| Populus euphratica | 497M | Salicaceae, Populus | Whole-genome shotgun |
| Amborella trichopoda | 748M | Amborellaceae, Amborella | Roche 454, Illumina |
Plant Genome Sequencing and Assembly
To date, comprehensive sequencing and assembly of several hundred plant genomes have been accomplished. These endeavors encompass a variety of model plants, cereal crops, horticultural species, oil crops, and bioenergy plants. In contrast to animal genomes, plant genomes exhibit significant complexities, characterized by highly repetitive sequences, transcription factors, retrotransposons, and polyploidy. These factors complicate the assembly and sequencing of plant genomes, introducing substantial uncertainty.
Advancements in sequencing technologies have substantially mitigated these challenges. The transition from Sanger sequencing to second-generation sequencing technologies—exemplified by Illumina and Roche 454 platforms—enabled de novo sequencing. Currently, third-generation single-molecule sequencing technologies, such as PacBio's Single Molecule Real-Time (SMRT) sequencing, continue to drive down costs while enhancing efficiency and accuracy.
Services you may interested in
Pre-Sequencing Preparation and Strategic Selection
Prior to commencing plant genome sequencing, it is essential to gather relevant species information and conduct a preliminary survey to assess the genome's complexity. This preliminary sequencing (survey sequencing) aims to determine the genome's size and heterozygosity. These factors critically influence the feasibility of advancing to subsequent sequencing phases. Typically, genomes with substantial size (exceeding 10 Gb) impose stringent demands on sequencing technologies, assembly software, and computational memory, thereby hindering successful assembly. Moreover, elevated heterozygosity may lead to an assembled genome that inaccurately exceeds the actual genome size.
If the heterozygosity of a species surpasses 0.5%, assembly may present significant challenges. Conversely, heterozygosity levels exceeding 1% render assembly exceedingly difficult, complicating subsequent biological analyses.
Given the variation in size and complexity of plant genomes, multiple critical factors must be meticulously considered when undertaking plant genome sequencing projects. Firstly, it is imperative to determine the sequencing technology to be employed and to establish the optimal length of the reads. Secondly, comprehensive genome coverage must be ensured, and the appropriate size of the library must be chosen judiciously. Moreover, suitable software should be selected for the assembly process. The strategy formulated at the inception of the study will have profound implications for the progress of genome completion; thus, selecting the appropriate sequencing method or platform is paramount.
At present, owing to the nascent stage of third-generation sequencing technologies, mainstream research methodologies primarily rely on first-generation and second-generation sequencing technologies. In this context, it is also necessary to construct libraries, such as BAC (Bacterial Artificial Chromosome), Fosmid, and Cosmid, and utilize sequencing with different grades of insert fragments. For species with smaller genomes, platforms such as Roche 454 or Illumina (formerly known as "Solexa") may be considered. Conversely, for complex large plant genomes, it is recommended to employ a combination of two or more sequencing platforms to facilitate more accurate genome assembly, thereby enabling the construction of either a scaffold-based or a high-resolution genome map.
Methods of Genome Assembly
The assembly of genomes constitutes an exceptionally intricate task, necessitating the processing of large-scale datasets generated by next-generation sequencing (NGS) technologies, often encompassing billions of reads. This process mandates the utilization of high-performance computing (HPC) servers. The selection of appropriate and efficient algorithms is crucial for the assembly of a substantial volume of reads. An optimal assembly methodology not only accelerates processing speed but also ensures the accuracy of the results. Currently, the primary genome assembly methodologies encompass three predominant techniques: the greedy algorithm, the overlap-layout-consensus (OLC) method, and the De Bruijn graph approach. Each of these methods possesses distinct characteristics and collectively contribute to the precise assembly of genomic data.
Annotation of Plant Genomes
Upon achieving predefined standards of completeness and contiguity in plant genome assembly, a comprehensive genomic sequence can be obtained. Subsequently, the application of bioinformatics methodologies and tools becomes essential for in-depth annotation of the plant genome. Despite variations in software algorithms utilized for different plant genomes, the annotation process generally encompasses four pivotal phases: prediction of repetitive sequences, identification of ncRNA, gene structure prediction, and functional annotation of genes. These steps collectively facilitate a thorough and systematic elucidation of the plant genome.
Prediction of Repetitive Sequences
In sequenced plant genomes, repetitive sequences often constitute a significant portion, frequently amounting to 50% or more of the entire genome. For instance, repetitive sequences in the soybean genome comprise 59%, whereas in the maize genome, they account for as much as 85%. Due to their low sequence conservation, repetitive sequences pose significant challenges for identification, necessitating the construction of a repetitive sequence database specific to the genome in question.
Current methodologies for predicting repetitive sequences for the purpose of genome annotation include three primary approaches: homology-based methods, de novo prediction techniques, and approaches utilizing cDNA expressed sequence tags (cDNA-ESTs). Commonly employed software tools in this context include ReASR,PFR-DF, and Piler.
Prediction of ncRNA
ncRNAs, which are RNA molecules that do not participate in protein translation, such as ribosomal RNA (rRNA) and transfer RNA (tRNA), are found in relatively low abundance within organisms. For instance, in Triticum aestivum (wheat), the length-to-quantity ratios of various ncRNAs are as follows: rRNA is 59.2 kb/328 molecules, tRNA is 187.4 kb/2585 molecules, microRNA (miRNA) is 47.5 kb/286 molecules, and snRNA is 14.9 kb/106 molecules. Despite representing a mere 0.01% of the entire genome, ncRNAs are indispensable for numerous biological functions.
Given the extensive variety and distinctive characteristics of ncRNAs, they lack the typical features of protein-coding genes. Consequently, research on ncRNAs predominantly focuses on predicting their stable secondary structures and sequence conservation. Tools such as the RNAstructure web server, and databases commonly used for ncRNA analysis, including RNAdb, NONCODE, Rfam, miRBase, and snolBase, are instrumental in these investigations. These methodologies enhance our understanding of the complex mechanisms by which ncRNAs operate within biological systems.
Gene Structure Prediction
The accurate prediction of gene structure facilitates the comprehensive acquisition of genomic distribution and structural information, thereby providing essential data for functional annotation and evolutionary analysis. This process encompasses the identification of specific gene loci, open reading frames (ORFs), translation initiation and termination sites, intron and exon regions, promoters, alternative splicing sites, and protein-coding sequences.
To ensure the precision and reliability of these predictions, a combination of sequence alignment and de novo prediction methodologies is employed. This integrative approach is supported by an array of specialized computational tools, including Genscan, Gene-MANIA, SNAP, Augustus, Climmer, ClimmerHMM, Glean, and EVidenceModeler. These tools collectively enable the meticulous characterization and analysis of the genomic architecture.
Functional Annotation of Genes
Upon obtaining the gene structure information, it becomes imperative to acquire comprehensive functional annotations. The functional annotation includes various aspects such as gene prediction, motif and domain prediction within the gene, and the annotation of protein function and associated biological pathways. Key databases utilized for functional annotation encompass the National Center for Biotechnology Information (NCBI), InterPro, SwissProt, Gene Ontology (GO), TrEMBL, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Clusters of Orthologous Groups (KOG/COG).
For functional annotation, homology-based approaches, such as Basic Local Alignment Search Tool (BLAST), are employed to identify homologous genes and annotate their functions accordingly. The UniProt protein sequence database was employed to acquire preliminary information concerning the sequences. The KEGG biological pathway database was utilized to predict the potential biological pathways in which the proteins, as well as associated metabolic processes, might be involved. The InterPro protein family database facilitated the identification of conserved sequences, motifs, and domains within the proteins. Furthermore, the GO functional annotation database was employed to predict the biological functions of the genes.
Evolutionary Analysis in Plant Comparative Genomics
Genomic traits, including size, internal arrangement, coding regions, and non-coding regions, exhibit a degree of variability. Comparative genomics, applied at the molecular level, can elucidate patterns of homogeneity and diversity unique to plant species. Comparative genomics entails the construction of maps using shared markers or sequencing corresponding genomic regions (or entire genomes) of different species. Analysis focuses on structural relationships, relative positions, and gene numbers to uncover the origins and functions of gene families and the mechanisms underlying their diversification and complexity through evolution.
Comparative genomics can be further classified into interspecific and intraspecific comparative genomics. Interspecific comparative genomics involves comparing the genomes of species with varying degrees of phylogenetic relatedness. Sequence alignment and analysis are employed to determine evolutionary relationships among species. Conversely, intraspecific comparative genomics examines genetic variability within a single species, often through resequencing studies. Such studies involve comparing sequences to a reference genome to identify patterns of SNPs and structural variation detection (SVD). This approach enhances the detection of genetic variation among individuals and contributes to the foundation of molecular breeding.
Services you may interested in
Summary
Preparation for Plant Genome Sequencing
With the advent of second-generation high-throughput sequencing technologies, alongside advancements in associated methodologies and assembly software, large-scale sequencing of plant genomes has been accelerating. Comprehensive analysis of plant genome sequences facilitates the acquisition of valuable information regarding genomic composition and functional gene sequences of the species in question, thereby providing a molecular basis for investigating plant molecular evolution, gene composition, and gene regulation. This approach also establishes a genetic foundation for the identification of important traits.
Given the substantial size and complex structure of plant genomes, characterized by a high degree of repetitive sequences and polyploidy, sequencing efforts are considerably challenged. Therefore, careful consideration is required in the selection of sequencing materials, analysis of sequencing results, and subsequent applications. Prior to large-scale sequencing, it is imperative to conduct a basic biological characterization of the target species, including assessments of heterozygosity, genome size, and chromosome ploidy.
Choosing an appropriate sequencing strategy demands an evaluation of the strengths and weaknesses of the available methods, allowing decisions based on species-specific genomic information. Employing a combination of sequencing approaches can enhance sequencing accuracy and facilitate effective data analysis. Upon obtaining sequencing data, it is crucial to employ suitable methods and software to assemble the data into a complete genome sequence. Concurrently, the construction of genetic or physical maps is indispensable, as they provide vital genetic background information for the species under investigation.
This structured approach to plant genome sequencing not only enhances the accuracy and reliability of genomic data but also supports subsequent genetic and functional analyses essential for advanced plant genomics research.
Analysis of Plant Genome Sequencing Results
Following extensive whole-genome sequencing and bioinformatics analyses, the resultant genomic sequences were subject to gene annotation. This step facilitated the identification and functional validation of genes associated with key phenotypic traits, such as flowering control, growth habits, dormancy, frost resistance, pest and disease resistance, fruit development, nutritional content, and quality attributes. Additionally, critical genes involved in secondary metabolite biosynthesis were also analyzed.
Furthermore, comparative genomics research can be employed to conduct in-depth synteny analyses between the genomic sequences of different plant species. This approach elucidates the origin and evolutionary relationships of plants while also exploring the influence of significant gene families on plant development.
Follow-up Research in Plant Genome Sequencing
The application of plant genome sequencing extends beyond the initial analyses discussed earlier. Subsequent research endeavors leveraging plant genomic data encompass several key areas:
Functional Genomics: Investigations involve the identification of novel genes, validation of their function through knockout strategies, and evolutionary analysis. These efforts aim to elucidate the interactions between plant growth, development, and environmental factors, thereby providing insights into the biological foundations of plant life.
Gene Expression Analysis: Using comprehensive genomic data, gene expression studies are conducted. Techniques such as Serial Analysis of Gene Expression (SAGE), cDNA microarrays, and DNA chips are employed to design gene-specific chips for expression profiling. Transcriptome sequencing of reference genomes is utilized to measure gene expression levels across different tissues or developmental stages, offering information on differential gene expression.
Genome Resequencing: Large-scale genome resequencing is undertaken to analyze genetic variation among different individuals or populations. This approach aims to identify genetic polymorphisms such as SNPs, InDels, andSVs. The derived genetic variation data are critical for understanding population-level genetic diversity.
Molecular Marker Development: The genome sequences provide a wealth of molecular markers, including simple sequence repeats (SSRs) and SNPs. These markers facilitate the construction of high-density genetic maps and physical maps, enhancing the localization of quantitative trait loci (QTLs).
Genome-Wide Association Studies (GWAS): GWAS employ linkage disequilibrium (LD) data from diverse populations and millions of SNPs across the genome. These studies conduct phenotype-genotype association analyses to identify genomic regions associated with target traits, thereby pinpointing genes correlated with rapid trait evolution and variation.
In conclusion, despite the inherent complexity of plant genomes, characterized by high repeat sequences and heterozygosity, significant progress has been made in sequencing these genomes. This advancement is primarily attributable to the development and application of high-throughput sequencing technologies, particularly second and third-generation sequencing methods. The increasing success in plant genome sequencing is evidenced by the substantial number of published articles in this domain annually. Additionally, some of the sequencing data continuously update plant genome databases, thereby providing a molecular basis for plant studies and offering new perspectives for species exploration. This progress has also accelerated biological research. Furthermore, the comparative analysis of certain genome sequences has facilitated the identification of numerous genes, thereby advancing our understanding of corresponding protein sequences and functions. It is anticipated that the ongoing development of sequencing technologies will resolve several outstanding issues in plant biology.